
Background
One of my papers involved calling somatic mutations in mouse tumors using next-generation sequencing (NGS) [1]. Figure 1 shows the final number of somatic mutations called per tumor. Although I did include all the necessary details behind my pipeline, I did not go into detail on how I chose the parameters I used. Normally, when you want to find mutations in a tumor you take a sample from normal (non-tumor) tissue in the same patient (mouse patient in my case). Then by comparing tumor and non-tumor tissue you can figure out what mutations are unique to the tumor (i.e. somatic mutations). However, the problem I encountered was most of my normal tissue samples were poor quality, thus making somatic mutation calling a bit less reliable. Sequencing coverage in most of the normal tissues was less overall and much more sporadic, so calling somatic mutations using each tumor’s paired normal sample will significantly underestimate the number of somatic mutations. There are numerous bioinformatics software packages out there for calling somatic mutations, and they all come with some default parameter settings which tend to do pretty well. However, in light of the challenges with my data I wanted some additional assurance that the parameters I chose work well.
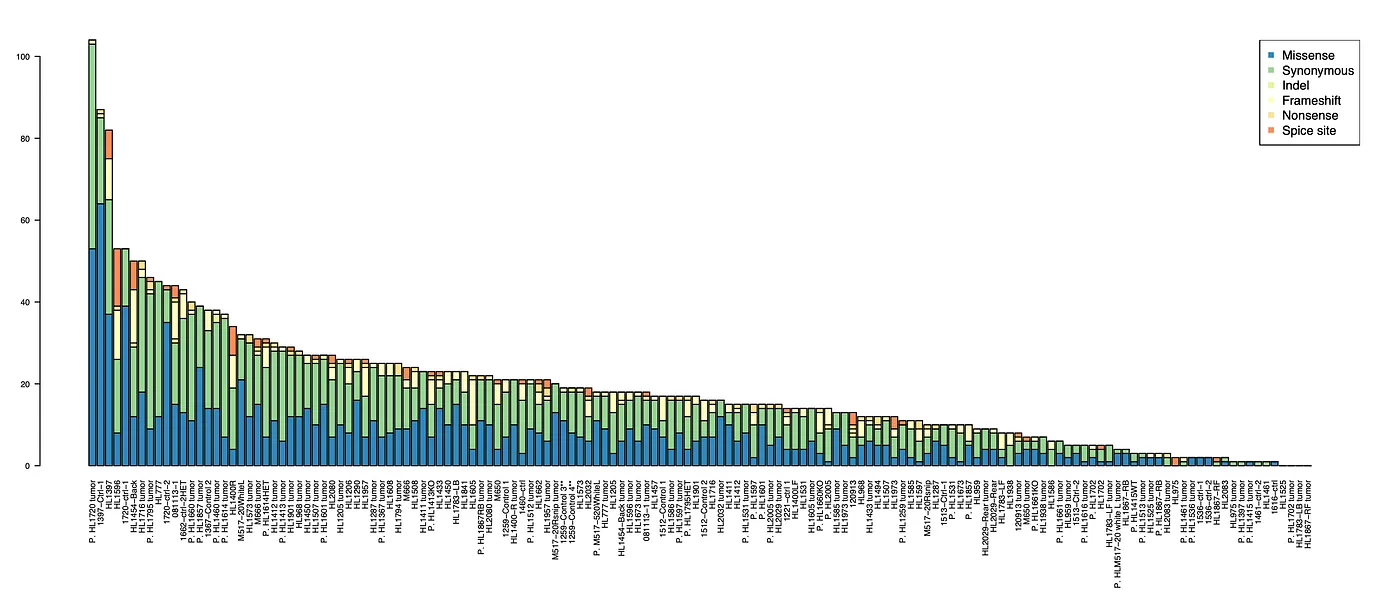
Figure 1: plots the number of mutations per tumor, broken down by type of mutation.
The problem with optimizing the parameters on my data is the total lack of training data. I had no way of knowing if a given somatic mutation is real or not. Although there are some “indicators” that tell me if a mutation is more likely to be true (e.g. if it has a high frequency in the tumor, the same mutation has been observed in other tumors, etc…), but I still don’t know if it real or not. However, I did come up with one reasonable indicator of what is likely a false positive mutation. To understand this you need to know about the dbSNP database. dbSNP characterizes genetic variation across populations by having assessed the allele status of millions of single nucleotide variants (SNPs). The primary dbSNP database is for humans, but there is also one for mice. Since the all the mice used in this study are of the same strain, if I find a SNP that is in some of the mice and is also in dbSNP, then I can reasonably assume the SNP is in all of the mice. Hence, these gives me a list of SNPs that if determined to be a somatic mutation in a tumor, it is likely a false positive.
Methods
The full bioinformatics pipeline used for the NGS data is detailed in my paper [1], but I’ll discuss the relevant parts for parameterizing the algorithm for identifying somatic mutation here:
Step 1 — Call initial mutation set My first step was to call an initial set of somatic mutations from all tumors using some lenient parameters. I am not worried about false positives here just yet.
Step 2 — dbSNP annotation I then determine whether each mutation exists in dbSNP.
Step 3 — Tumor and control sample pileups Combine all the somatic mutation calls and obtain pileups for each sample using the combined data. A pileup is a file that contains the total sequencing read coverage at each mutation site and the number of reads that support the mutation versus reference allele.
Step 4 — Parameter grid search Use the pileup files to call somatic mutations in each sample. False positive somatic mutations are defined as those which are found in dbSNP and are found in at least two normal samples. Calling somatic mutations is done with every combination of parameters listed in

Table 1: parameters benchmarked.
Results
Figures 2 and 3 plot the number of somatic mutations identified with various parameter values. The parameter that had the biggest impact on the number of somatic mutations called was Tn, which controls the maximum number of tumors a mutation can be called in before removing it as a false positive. The other parameters had significantly less effect. Similarly, Figures 4 and 5 show the proportion of false positive somatic mutations with respect to various parameter values. Recall that a false positive mutations is defined as any mutation that exists in dbSNP and occurs in two or more normal samples. Again, Tn had by far the biggest impact on the proportion of false positives.
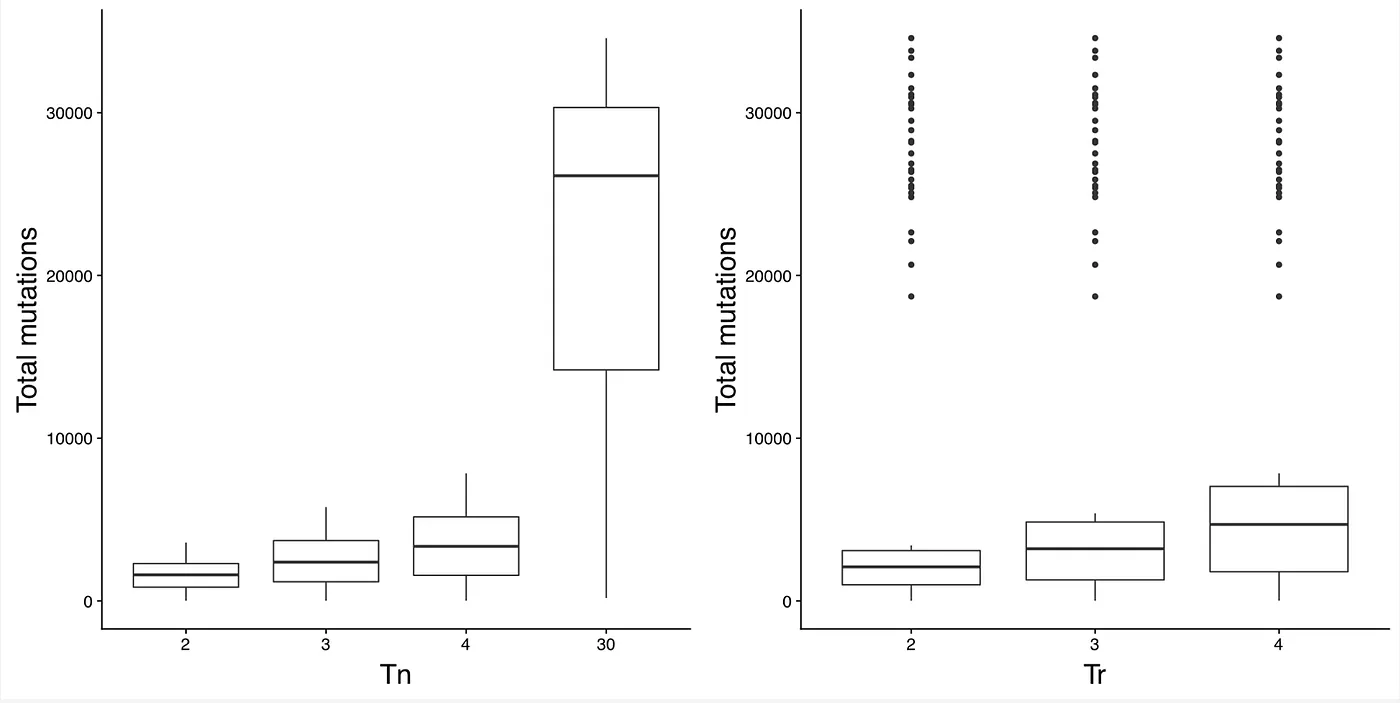
Figure 2: Number of total somatic mutations with different values of Tn and Tr.
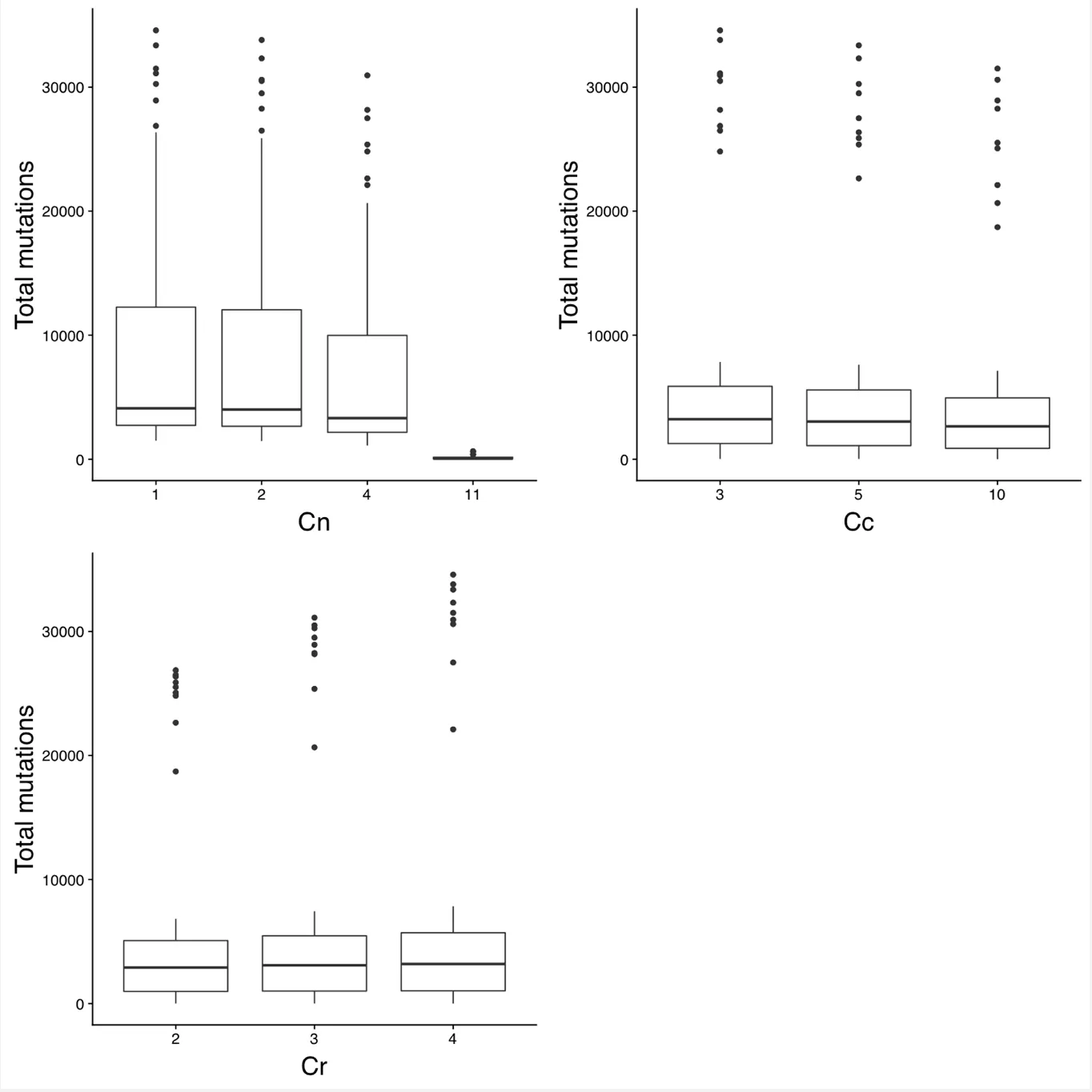
Figure 3: Number of total somatic mutations with different values of Cn, CCc, and Cr.
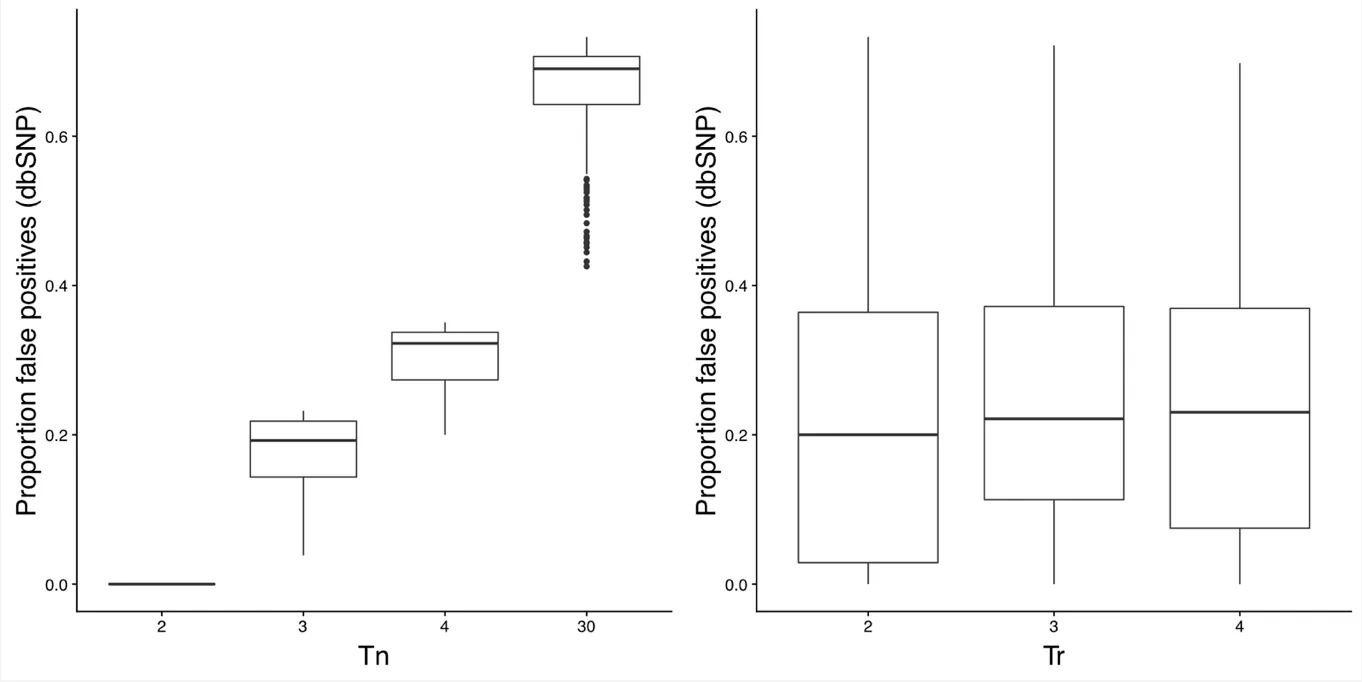
Figure 4: Proportion of false positive somatic mutations with different values of Tn and Tr.
Figure 5: Proportion of false positive somatic mutations with different values of Cn, CCc, and Cr.
Conclusions
There were two main challenges in identfying somatic mutations in my tumor dataset:
The normal control samples were poor quality and most could not be used for calling somatic mutations. This causes problems when using standard bioinformatics software which rely on having a good quality normal control sample for each tumor. I did not have a training dataset to parameterize my somatic mutation calling pipeline. My solution to the above two problems was to come up with a metric to estimate the number of false positives. This was done by assuming a somatic mutation is a false positive if it meets two conditions: (1) is is found in the dbSNP database and (2) it is found in two or more normal control samples. Such mutations are then assumed to exist in all samples and, hence, is not a somatic mutation.
Five different parameters were varied for calling somatic mutations. The parameter that had the biggest impact on the number of somatic mutations called was Tn, which controls the maximum number of tumors a mutation can be called in before removing it as a false positive. The remaining parameters had minimal impact. The parameters I ended up using in my paper where as follows: Tr=4, Cn=1, Cc=12, and Cr=4. However, as discussed in the supplementary methods of my paper, I ended up not using Tn to remove mutations. I instead used the Fisher’s exact test for removing mutations that occur in too many tumor samples.
References
- Liu H, Murphy CJ, Karreth FA, Emdal KB, Yang K, White FM, et al. Identifying and Targeting Sporadic Oncogenic Genetic Aberrations in Mouse Models of Triple-Negative Breast Cancer. Cancer Discovery. 2017 Dec 4;8(3):354–69.